Recent
Recent
Photo 29475566 © Jarek2313 | Dreamstime.com

Photo 167081387 © Tulipdesign18 | Dreamstime.com

Top Stories
Top Stories
(Image credit: Harvard SEAS)

Recommended
Recommended
Read This Next
Read This Next
© Fraunhofer ILT, Aachen, Germany/Andreas Steindl

Photo 301002215 © Artitwpd | Dreamstime.com

(Image credit: Jerzy Szuniewicz)

Learning Resources
Learning Resources
Courtesy of Shanghai Optics Inc





Sponsored Content
(Image credit: Palashuddin Sk)

(Photocollage created by Jacob Deats, Laboratory for Laser Energetics)

(Image credit: Jiaye Chen)

Photo 21585186 © Image191 | Dreamstime.com

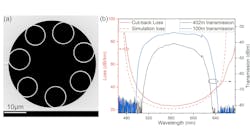
(Photo credit: Vescent)

(Photo credit: Quantum Science)


(Photo credit: QinetiQ)

(Image credit: VueReal)

Photo 305702414 © Zolak Zolak | Dreamstime.com

(Image credit: Focuslight Technologies/Susanne Westenhoefer)



(Image credit: Duke University)

(Image credit: Mikhail Volkov/University of Konstanz)

(Photo credit: ZEISS)

(Image credit: Endeavor Business Media)


(Photo credit: Quantum Corridor)

(Image credit: Duma Optronics)
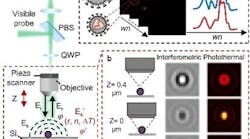
(Image credit: Ji-Xin Cheng’s Lab, Boston University)




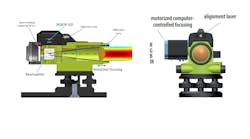
(Image credit: TRUMPF Scientific Lasers)


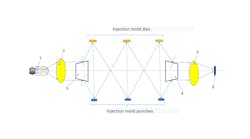

(Image credit: EPFL/Gözden Torun)

Photo 131219064 © Photoking | Dreamstime.com

Photo 174814076 © Artur Szczybylo | Dreamstime.com

Photo 11144854 © Mark Herreid | Dreamstime.com

(Image credit: GQI)
Photo 5322337 © Demarco | Dreamstime.com

(Image credit: Endeavor Business Media)


(Courtesy of JJ Coello-Bravo)

Featured Media
Featured Media

Home
Photonics Hot List: April 26, 2024
April 26, 2024

Home
Photonics Hot List: April 19, 2024
April 19, 2024

Home
Photonics Hot List: April 12, 2024
April 12, 2024

Leadership in Lasers and Photonics
Welcome to Laser Focus World's Leadership in Lasers and Photonics program.